 |
 |
|
|
 Форум судебных медиков России > Судебная медицина и судебно-медицинская экспертиза > Аналитическая и судебная токсикология
Форум судебных медиков России > Судебная медицина и судебно-медицинская экспертиза > Аналитическая и судебная токсикология  |

 |
Предел детектирования и количественного определения, LOD& LLQ |
 |
| vea |
 20.03.2013 - 20:39 20.03.2013 - 20:39
Сообщение
#16 |
|
|
Спасибо всем. Попробую применять. Но что-то меня смущает. Я не великий математик, но возможности выбрать необходимый мне способ апроксимации я не увидела: авто и другое. Мне не понятно. Если не трудно, поясните. Буду благодарна.
|

  |
  
|
 |
 |
| Deminolog |
20.03.2013 - 21:23
Сообщение
#17 |
|
|
В приложенном файле скриншот настроек линии тренда. Для начала надо определиться, какая функция описывает лучше Вашу кривую. Полиномиальная, линейная, экспонента... Вот и все
 А название аппроксимирующей - это всего-навсего то, как она будет названа в легенде графика А название аппроксимирующей - это всего-навсего то, как она будет названа в легенде графика Эскизы прикрепленных изображений 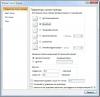
|
|
|
|
|
|
| ws_K |
21.03.2013 - 12:33
Сообщение
#18 |
|
|
Уважаемые коллеги.
Сравнительно недавно (1993-1994 г.г.) я имел прямое отношение к разработке «Автоматизированной системы массовых хроматографических исследований крови, мочи и вытяжки мягких тканей на содержание алкоголя и его суррогатов». Пользуясь этим обстоятельством, а также тем, что в процессе разработки был накоплен некоторый опыт, попробую внести ясность в обсуждаемый вопрос. Вынужден извиниться – движок Форума не позволяет отражать математические выражения адекватно. Поэтому основной текст сообщения в прикрепленном файле Прошу учесть также – изложение касается методологии газо-жидкостной хроматографии, но с легкостью может быть распространено на иные методологии, в основе которых лежат сравнения с «эталоном».  _______.doc ( 61.5 килобайт )
Кол-во скачиваний: 787
_______.doc ( 61.5 килобайт )
Кол-во скачиваний: 787С уважением, ws_K. |
|
|
|
|
|
| Deminolog |
21.03.2013 - 15:24
Сообщение
#19 |
|
|
Прочитал.
Уважаемый ws_K! Я, конечно, не имею таких заслуг, однако мне не совсем понятно, что Вы имеете в виду в ряде моментов, поэтому предлагаю обсудить данный вопрос. Если результат не попадает в градуировку - поправочные коэффициенты Вас тут не спасут (готов принять исключением тот вариант, когда градуировка на чистых соединениях, без учета матрицы, и то, считаю что это не совсем корректно). Про 10 сигма - это личное дело каждого. Хотите - берите 3, 6... Вариантов-то много. Главное - чтобы Вы были уверены, что это именно Ваш сигнал, а не шум. В случае 10 сигма - это не "хрен с ним, будь что будет", это значит, что сигнал, который Вы регистрируете в 10 раз превышает фоновый. А вот теоретический расчет пределов обнаружения и детектирования - вот это действительно пальцем в небо, хотя опытный специалист может не так уж сильно ошибиться. Далее, что Вы понимаете под динамической характеристикой хроматографа? Динамический диапазон? Тогда уж не хроматографа, а детектора, если быть совсем точным. Если Вы имеете в виду работу в участке, близком к середине градуировочного графика - это одно. Если что-то другое - пожалуйста, поясните. В первом случае - я согласен с Вами. Но опять-таки, Вы работаете в границах диапазона линейности. Далее, о коэффициентах. Знаете, у меня сложилось впечатление, что Вы говорите о матричном влиянии. Если это так - то это я еще как-то понимаю, если нет - не понимаю вовсе. Классическая процедура для работы со сложными объектами - использование бланка, холостого, в который вносят уже стандартную добавку. Сравнивая сигнал со стандартной добавки и без оной Вы оцениваете мешающие влияния, которые легко могут доходить до 70 и даже более %. Прикладываю еще одну достаточно полезную статью, пусть и не самую свежую, но опираться на неё можно. Это перевод рекомендаций ИЮПАК. Прикрепленные файлы  _____________________________________________.pdf ( 2.07 мегабайт )
Кол-во скачиваний: 625
_____________________________________________.pdf ( 2.07 мегабайт )
Кол-во скачиваний: 625 |
|
|
|
|
|
| Deminolog |
21.03.2013 - 15:39
Сообщение
#20 |
|
|
И, да, по поводу того, что диапазон линейности устанавливается следует понимать следующим образом: Вы действительно вводите последовательно разные концентрации определяемых веществ, после чего строит зависимость аналитического сигнала от концентрации, но Вы всегда должны проводить следующую работу: оценивать, где тот самый диапазон линейности. В какой-то момент график вообще может на плато выйти (например, превышена максимальная величина оптической плотности для спектрофотометра), но перед этим Вы будете наблюдать существенные отклонения от основного закона светопоглощения. В диапазоне низких концентраций также возможны отклонения. Надо оценивать коэффициенты, смотреть разброс, делать повторности, смотреть воспроизводимость вкола, воспроизводимость результата. Или Вы с этим не согласны?
|
|
|
|
|
|
| D'ng |
21.03.2013 - 16:00
Сообщение
#21 |
|
|
Здравствуйте.
Ув. ws_K постарайтесь все таки писать в сообщении , а не в прикрепленных файлах. В дополнении к отмеченному Deminolog-ом Расчет результатов по калибровочному графику подразумевает работу именно в диапазоне концентраций вашего графика. Применять поправочные коэффициенты для расчета образцов , "выскочивших" за рабочий диапазон не верно. Для таких проб применяется разбавление (высокие концентрации) или стандартная добавка определяемого вещества в пробу (для низких). Применительно же к этанолу то искать сотые доли промилле смысла нет. ПС... Исправил сообщение. Не десятые , а сотые доли промилле. Сории за описку. |
|
|
|
|
|
| KNIISE |
22.03.2013 - 13:04
Сообщение
#22 |
|
|
Следует помнить: динамический диапазон детектора и хроматографической системы в конкретной конфигурации - вещи разные.
|
|
|
|
|
|
| ws_K |
25.03.2013 - 17:33
Сообщение
#23 |
|
|
Цитата(Deminolog @ 21.03.2013 - 15:24)  Я, конечно, не имею таких заслуг... Ничего, все впереди!Цитата(Deminolog @ 21.03.2013 - 15:24) ...поправочные коэффициенты Вас тут не спасут... Да я на их помощь и не рассчитываю: спасение утопающих – дело рук самих утопающих.Но, если серьезно. Какие «поправочные» коэффициенты? Если Вы имеете ввиду коэффициенты a и b функции калибровки системы y = ax + b, то это не «поправочные», а «фундаментальные» коэффициенты, полученные в результате калибровки хроматографической системы. Разве Вы не используете функцию калибровки в своих хроматографических исследованиях? Если – нет, то, как это?.. Именно об этой функции и об этих же коэффициентах идет речь в п. 6.2.1. упомянутого ГОСТа. Отличия в том, что в ГОСТе они имеют обозначения бета 1 и бета 0, соответственно, а для оценки линейности этой функции, используется статистика т.н. остатков; в моем сообщении для этих же целей используется другая статистика - коэффициент корреляции r, и показан (пошагово) способ его «ручного» расчета. О функции калибровки идет речь и на стр. 104 Рекомендаций IUPAC (еще раз, спасибо Вам за полезный источник). Там, правда, «свои» термины и обозначения, зато упомянут коэффициент корреляции. Цитата(Deminolog @ 21.03.2013 - 15:24) ...что Вы понимаете под динамической характеристикой хроматографа? Под динамической характеристикой любой измерительной системы я понимаю, как и подавляющее большинство лиц, связанных с измерениями, функциональную зависимость между откликом системы и входным воздействием. Применительно к хроматографии: входным воздействием является отношение высоты пика исследуемого вещества к высоте пика внутреннего стандарта; откликом является измеренная концентрация исследуемого вещества; отклик системы прямо пропорционален входному воздействию. Вы понимаете под этим что-то иное? Динамический диапазон указывает min. и max. значения, между которыми может изменяться отклик; Динамическая характеристика – это функция, указывающая, как именно меняется отклик в пределах динамического диапазона. Применительно к хроматографии эта функция должна быть линейна, но в различных приборах (например, фотометрах), динамическая характеристика может быть логарифмической или, напротив, экспоненциальной. Зная динамическую характеристику системы легко определить ее динамический диапазон. Динамический диапазон детектора и хроматографической системы в целом, как верно отметил уважаемый KNIISE, могут существенно различаться. Дело в том, что источников нелинейности в хроматографической системе несколько: детектор, усилитель сигнала детектора, регистратор. В «алкогольной» задаче обычно используют катарометр – детектор по теплопроводности. В интересующем нас диапазоне (от 0 до 6.0 промилле), и даже существенно выше, абсолютное большинство катарометров линейны. Регистраторы (как правило – потенциометры постоянного тока) в пределах своей шкалы, также линейны. Основным источником нелинейности является усилитель постоянного тока. Если сигнал на его входе окажется слишком велик, он может попасть в т.н. «область насыщения» усилителя, что приведет к нелинейности динамической характеристики системы. Чтобы этого не происходило, на входе усилителя имеется делитель, с помощью которого входной сигнал ослабляют до разумного уровня. Но самым-самым главным источником нелинейности в хроматографической системе является четвертый ее «компонент» - человек. Обычно это лаборант, подготавливающий калибровочные смеси (т.н. «график»). Представьте себе, что в одном из флаконов нарушено «паспортное» соотношение концентраций EtOH и i-PrOH (рука дрогнула, например). И все. Нелинейность системы обеспечена. При калибровке хроматографической системы мы с Вами оцениваем нелинейность системы в целом, а не отдельных ее компонент. Цитата(Deminolog @ 21.03.2013 - 15:24) Далее, о коэффициентах... По поводу коэффициентов, я, вроде бы, все пояснил, а о «мешающих влияниях» я не говорил вовсе. В «алкогольной» задаче таких влияний попросту нет. В период с 1993 года и по сей день, вблизи пиков EtOH и i-PrOH никаких иных пиков я, что-то, не наблюдал. Цитата(Deminolog @ 21.03.2013 - 15:24) [фразу о том, что] диапазон линейности устанавливается, следует понимать следующим образом... Вашу мысль я понял, но вот вопрос: не слишком ли трудоемко «устанавливать»? Нет-нет, я двумя руками «за» определение концентрации вещества по нескольким измерениям, но не слишком ли много «эвристики» в Вашем подходе? Чтобы оценивать коэффициенты, разброс, воспроизводимость нужно иметь четкие критерии. Если они есть, не составляет труда формализовать оценки и придти к определенным, а не «эвристическим» выводам. Например, о линейности системы можно судить по величине коэффициента корреляции: если r < 0.98 – динамическая характеристика нелинейна. Если r < 0.98 и Вас интересует концентрация, с которой проявляется нелинейность, нужно отбросить одну из граничных (как правило, верхнюю) точку калибровочной шкалы, и заново рассчитать r. Если заново вычисленное значение r окажется больше и равным 0.98 – динамическая характеристика нелинейна на участке между предпоследней и последней точками «графика», и т.д. А вот как, например, оценивается воспроизводимость при калибровке в нашей автоматизированной системе: По трем вкалываниям проб из трех разных флаконов с соответствующей концентрацией EtOH вычисляют среднее значение отношения высот пиков EtOH/i-PrOH и ошибку среднего. Если ошибка среднего превышает ±5% - воспроизводимость признают неудовлетворительной. В упомянутом ГОСТе приведен другой способ оценки воспроизводимости. А как оцениваете воспроизводимость Вы? Уважаемый D'ng. Не могу гарантировать Вам, что впредь не буду использовать прикрепленные файлы с сообщениями. Поверьте, это не блажь, а вынужденная необходимость. Причины я объяснил в предыдущем сообщении. Но Вы – свободный человек в свободной стране, и можете свободно НЕ читать прикрепленные файлы. Не правда ли? Цитата(D'ng @ 21.03.2013) Расчет результатов по калибровочному графику подразумевает работу именно в диапазоне концентраций вашего графика. Отнюдь. Этим утверждением Вы лишаете функцию калибровки экстраполирующих свойств, а и без того несчастное Человечество – возможности заглянуть в Неизвестное. Единственное, что необходимо сделать в случае, если отсчет «вылетел» за пределы калибровочной шкалы – добавить его в «график» как конечную точку калибровочной шкалы и заново вычислить r. Если его значение по-прежнему будет больше или равно 0.98 – все в порядке, Вы все еще находитесь на линейном участке динамической характеристики системы; если оно станет меньше 0.98 – Вы можете быть уверенны в том, что угодили на нелинейный участок характеристики, и результату доверять нельзя. О «поправочных» коэффициентах – см. выше. Ниже Вы рассказали о приеме, который, действительно, широко применяется в практике хроматографических исследований. Однако я никогда не видел, чтобы он применялся в «алкогольных» исследованиях: нет необходимости (концентрации этилнитрита превышающие 6.0 промилле в объектах практически не встречаются, а если и встречаются, то обусловлены, как правило, причинами, не связанными с потреблением алкоголя «реципиентом»). Цитата(D'ng @ 21.03.2013) Применительно же к этанолу то искать сотые доли промилле смысла нет. Ну, почему же? Знаете, есть такой раздел математики: «Теория ошибок». К сожалению, судебные медики им часто пренебрегают. Между тем, один из его постулатов можно свести к фразе: «Хочешь быть уверенным в первом знаке после десятичной точки – вычисляй (измеряй) с точностью до второго». Показывать или нет второй знак – вопрос вкуса эксперта, но возможность правильно указать первый, у него, при этом, сохраняется. Вообще же, последний знак после точки характеризует не столь количество чего-либо, сколь точность, с которой произведены измерения или вычисления. С уважением, ws_K. |
|
|
|
|
|
| Deminolog |
25.03.2013 - 21:48
Сообщение
#24 |
|
|
Трижды "ой"!
Вы не против, если я не буду цитировать отдельные моменты, а просто продолжу дискуссию в повествовательном русле? Поправочные коэффициенты не относятся к уравнению регрессии. Они относятся к результату, учет определенных влияний на конечный результат, так сказать. По поводу коэффициента В: его значимость или не значимость в уравнении регрессии устанавливается отдельно, это довольно муторная процедура, надо признать. Понятия динамической характеристики хроматографа я не видел вообще, хотя не первый год варюсь в этом котле, откровенно говоря. Но я бы понял это немного иначе. Например, как набор определенных технических характеристик хроматографа, но никак не отношение к детектированию. Я не претендую на истину в последней инстанции, но и сказать, что ничего в этом не понимаю тоже не могу Немного тяжеловато будет дальше, поскольку некоторые вещи уже давно являются аксиомами в аналитике... Например, никогда не давать оценку результата, который не вписывается в градуировку Каким бы ни был линейным детектор, обязательно проводить исследование всего диапазона, поскольку даже малейшие изменения, погрешности, которые будут вноситься, скажутся на коэффициентах уравнения.Мои критерии к воспроизводимости результата, в принципе, сопоставимы. В частности, при n = 6 (где n - число повторностей, а каждая повторность состоит из полного набора точек градуировки, которые откалываются по 3 раза каждая) я также считаю значимым отличие более 5%. Далее, почему я против (категорически) экстраполяции... Вот Вы привели в качестве примера катарометр. Вообще, я считаю и катарометр, и ПИД линейными детекторами, но при этом все равно необходимо линейность показывать и доказывать. Таковы правила игры. Но теперь отойдем от этих двух детекторов, возьмем еще один вполне распространенный... Спектрофотометрический. И, даже, пожалуй, масс-спектрометрический. Вот тут-то и наступает веселая жизнь. Об отклонениях от основного закона светопоглощения я, пожалуй, писать не стану, в учебниках по аналитической химии Золотова, Кельнера, Отто это все прекрасно написано, но в случае чего готов вставить сюда выдержки. Также как и не стану писать о тонкостях калибровки на масс-спектрометрах (тут уже вообще целую тему открывать можно, с некоторыми коллегами мы этот вопрос обсуждали уже). Просто создается впечатление, что Вы предлагаете чуть ли не ограничиться 2-мя точками и все Ведь, как известно, через 2 точки можно провести прямую и только одну. Результат достоверен только в том случае, если диапазон концентраций, в котором он находится, вписывается в градуировочную зависимость.Также не стоит забывать о влияниях условий хроматографирования. Я могу добиться лучших пределов детектирования изменением условий хроматографирования. Если пик стал уже, он стал выше, а значит на меньших концентрациях я все равно буду видеть его уверенней. Вот как можно дать количественную оценку сигналу, который едва выбивается из шума? Точнее, не превосходит фоновый сигнал хотя бы в 3 раза. Вы сможете поручиться, что это сигнал? Я - нет. Сумбруно, наверное получилось, да и не на все моменты ответил, но прошу меня извинить, вечером голова уже не очень варит, чтобы отвечать хорошо и емко... С уважением, А. |
|
|
|
|
|
| D'ng |
25.03.2013 - 23:15
Сообщение
#25 |
|
|
Цитата(ws_K @ 25.03.2013 - 16:33) Не могу гарантировать Вам, что впредь не буду использовать прикрепленные файлы с сообщениями. Поверьте, это не блажь, а вынужденная необходимость. Причины я объяснил в предыдущем сообщении. Но Вы – свободный человек в свободной стране, и можете свободно НЕ читать прикрепленные файлы. Не правда ли? Отнюдь. Этим утверждением Вы лишаете функцию калибровки экстраполирующих свойств, а и без того несчастное Человечество – возможности заглянуть в Неизвестное. Единственное, что необходимо сделать в случае, если отсчет «вылетел» за пределы калибровочной шкалы – добавить его в «график» как конечную точку калибровочной шкалы и заново вычислить r. Если его значение по-прежнему будет больше или равно 0.98 – все в порядке, Вы все еще находитесь на линейном участке динамической характеристики системы; если оно станет меньше 0.98 – Вы можете быть уверенны в том, что угодили на нелинейный участок характеристики, и результату доверять нельзя. О «поправочных» коэффициентах – см. выше. Ниже Вы рассказали о приеме, который, действительно, широко применяется в практике хроматографических исследований. Однако я никогда не видел, чтобы он применялся в «алкогольных» исследованиях: нет необходимости (концентрации этилнитрита превышающие 6.0 промилле в объектах практически не встречаются, а если и встречаются, то обусловлены, как правило, причинами, не связанными с потреблением алкоголя «реципиентом»). Ну, почему же? Знаете, есть такой раздел математики: «Теория ошибок». К сожалению, судебные медики им часто пренебрегают. Между тем, один из его постулатов можно свести к фразе: «Хочешь быть уверенным в первом знаке после десятичной точки – вычисляй (измеряй) с точностью до второго». Показывать или нет второй знак – вопрос вкуса эксперта, но возможность правильно указать первый, у него, при этом, сохраняется. Вообще же, последний знак после точки характеризует не столь количество чего-либо, сколь точность, с которой произведены измерения или вычисления. С уважением, ws_K. По поводу обсуждений в прикрепленных файлах... Теперь это не просьба , а настоятельная рекомендация. В файлах выкладывайте только то , что не можете написать в сообщении .Примите к сведению. По поводу калибровочных мы не обсуждаем возможность человечества заглянуть в "неизвестное". Если ваша проба вылетела за диапазон калибровочной то , что вы добавляете в график калибровочной ? Новый стандарт , соответствующий концентрации пробы? Вопрос целесообразности никто не снимал с обсуждения. (калибровка до 12 промилле по этанолу в крови для вас новость ?) По поводу низких концентраций этанола... Беда России - дураки во власти. Если вернут допустимую концентрацию этанола то не важно 0,11 или 0,105. Более того из личного опыта . любители выдать результат 0,1245 промилле алкоголя в крови обычно садятся в лужу при вопросе о повторном замере в том же образце. |
|
|
|
|
|
| ws_K |
25.03.2013 - 23:41
Сообщение
#26 |
|
|
Цитата(Deminolog @ 25.03.2013 - 21:48) Вы не против, если я не буду цитировать отдельные моменты, а просто продолжу дискуссию в повествовательном русле? Конечно, не против. Т ем более, что дискуссия, во всяком случае, пока, носит конструктивный характер. Мне не хотелось бы «влет» реагировать на Ваше сообщение – надо подумать. Ладно?С уважением, ws_K. |
|
|
|
|
|
| ws_K |
26.03.2013 - 00:32
Сообщение
#27 |
|
|
Уважаемый D'ng.
Я не увидел конструктива в Вашем последнем сообщении – одни амбиции, отсутствие чувства юмора и немотивированная агрессивность… Право, не стал бы отвечать, но, что поделаешь, природное любопытство. Калибровка в диапазоне до 12 промилле по этанолу в крови для меня, действительно, новость. Нельзя ли ссылку, хотя бы библиографическую, на документ (Методическое письмо, Методические рекомендации, проч.), устанавливающий эту норму в практике медико-токсикологических отделений БСМЭ? С уважением, ws_K. |
|
|
|
|
|
| D'ng |
26.03.2013 - 00:37
Сообщение
#28 |
|
|
Ув. ws_K постарайтесь не сводить свои сообщения до уровня флуда.
|
|
|
|
|
|
| ws_K |
1.04.2013 - 01:04
Сообщение
#29 |
|
|
Уважаемый Deminolog.
Я уверен в том, что Вы – хороший и знающий специалист. Именно поэтому для меня представляет определенный интерес Ваша точка зрения. И все-таки, я не могу понять, о каких «поправочных» коэффициентах Вы говорите. В моем сообщении есть единственный поправочный коэффициент k, связывающий обнаруженную концентрацию этанола с конкретным объектом исследования: кровью, мочой или с вытяжкой из мягких тканей. Если Вы имеете в виду его, то должен сказать: этот коэффициент не моя «придумка», он не предназначен для спасения кого-либо от чего-либо. Это требование медико-токсикологической Методики, от которой Вы, как судебный медик-токсиколог, отступать не имеете права. Требования Методики обоснованы: истинная концентрация этанола зависит не только от соотношения пиков на хроматограмме, но и от физико-химических свойств исследуемого объекта. Никаких иных функций, связанных с линейностью, экстраполяцией, воспроизводимостью, проч. этот коэффициент не несет. Я угадал - мы так долго говорили о k? Ответьте, пожалуйста – спать не буду! Цитата(Deminolog @ 25.03.2013 - 21:48) По поводу коэффициента В: его значимость или не значимость в уравнении регрессии устанавливается отдельно, это довольно муторная процедура… Это я не берусь комментировать, поскольку не понимаю, о чем идет речь. Давайте еще раз запишем уравнение регрессии: y = ax + b. Если Вы употребляете слово «значимость» как термин математической статистики – тогда Вы употребляете его не по назначению: и коэффициент регрессии a (иногда называют угловым коэффициентом), и свободный член b (иногда называют коэффициентом сдвига) определяют по выборке. Они всегда значимы (даже если равны нулю), поскольку, совместно, полностью определяют линейную регрессию по калибровочной выборке. Просто при b = 0 линия регрессии проходит точно через начала координат. Возможно, Вы имели в виду определение доверительных пределов коэффициентов a и b (точность, с которой они вычислены). Для этого используют формулу Бартлета, в которую входит и коэффициент корреляции r. Процедура, действительно, очень громоздкая, и мало кто ее применяет на практике. Цитата(Deminolog @ 25.03.2013 - 21:48) Понятия динамической характеристики хроматографа я не видел вообще… Любой аналоговый измерительный прибор (хроматограф, в частности) полностью описывается двумя характеристиками: амплитудно-частотной (АЧХ) и динамической (ДХ). Первая отражает реакцию прибора на скоростнЫе, а вторая – на амплитудные изменения входного сигнала. В том случае, если АЧХ плоская во всем диапазоне временнЫх изменений сигнала, а ДХ линейна во всем диапазоне изменений его амплитуд, сигнал передается без искажений формы и может быть измерен с достаточно высокой точностью. Каждый раз, когда мы с Вами вкалываем в испарительную камеру смесь спиртов с целью определить времена выхода соответствующих им пиков (качественная калибровка), мы исследуем, в сущности, АЧХ хроматографа; когда мы вкалываем набор смесей спирта в соответствующей концентрации и внутреннего стандарта (количественная калибровка), мы исследуем ДХ хроматографа. Динамическая характеристика имеет множество синонимов: например в радиоэлектронике ей соответствует термин «передаточная функция». Цитата(Deminolog @ 25.03.2013 - 21:48) Понятие динамической характеристики хроматографа… я бы понял немного иначе. Например, как набор определенных технических характеристик хроматографа, но никак не отношение к детектированию. Вы знаете, по-моему, только две технические характеристики – масса и габариты хроматографа, не имеют отношения к детектированию. Все остальное, от материала колонки, до скорости протяжки ленты самописца к детектированию имеет самое прямое отношение. Цитата(Deminolog @ 25.03.2013 - 21:48) …я против (категорически) экстраполяции... Вообще, я считаю и катарометр, и ПИД линейными детекторами, но при этом все равно необходимо линейность показывать и доказывать. Таковы правила игры... Я тоже считаю эти детекторы линейными в широком диапазоне концентраций. А по поводу игр… Вот выдержки из двух документов, регламентирующих «алкогольные» исследования – 1. Методические рекомендации МОЗ Украины. Киев, 2006 г.: «Качественная калибровка выполняется по смеси спиртов, содержащей MeOH, EtOH, i-PrOH, PrOH, i-BuOH, BuOH, i-AmOH, AmOH». «Количественная калибровка выполняется по смеси спиртов EtOH и i-PrOH. Градуировочная шкала: 0.25, 0.50, 1.0, 2.0, 4.0, 5.0 промилле EtOH; 4.0 промилле i-PrOH». 2. Методические указания. Москва, 1967 г.: Степень выраженности алкогольного опьянения. (Прозоровский, Карандаев, Рубцов). менее 0,3‰ – отсутствие влияния алкоголя; от 0,3 до 0,5‰ – незначительное влияние алкоголя; от 0.5 до 1,5‰ – легкое опьянение; от 1,5 до 2,5‰ – опьянение средней степени; от 2,5 до 3,0‰ – сильное опьянение; от 3,0 до 5,0‰ – тяжелое отравление алкоголем, может наступить смерть; от 5,0 до 6,0‰ – смертельное отравление. Так вот, применяя эти два документа совместно, и опираясь на Вашу логику, установить «смертельное отравление» нельзя, поскольку диапазон 5.0 – 6.0 промилле лежит за пределами калибровочной шкалы, а менять калибровочную шкалу невозможно, поскольку это отступление от методики. Как бы поступили Вы в этой "игре"? Цитата(Deminolog @ 25.03.2013 - 21:48) …отойдем от этих двух детекторов [катарометр, пламенно-ионизационный], возьмем еще один – спектрофотометрический... или масс-спектрометрический. Я не могу об этом судить: спектрофотомерией и масс-спектрометрией не занимался, но допускаю, что там проблем не меньше, чем в хроматографии. Цитата(Deminolog @ 25.03.2013 - 21:48) Просто создается впечатление, что Вы предлагаете, чуть ли не ограничиться 2-мя точками [при калибровке] и все. Это - впечатление. Правда, я не понял, на чем оно основано? Отступления от методики (см. выше) я считаю недопустимыми, и о калибровке хроматографа по двум точкам, я не говорил, и говорить не мог в принципе. Цитата(Deminolog @ 25.03.2013 - 21:48) …как можно дать количественную оценку сигналу, который едва выбивается из шума? Точнее, не превосходит фоновый сигнал хотя бы в 3 раза. Мне не очень понятен практический смысл такой оценки. Положим для определенности (числовой пример реальный): мы хотим оценить концентрацию этанола в крови; высота пика этанола на хроматограмме равна утроенной толщине изолинии (т.е., сигнал в три раза превосходит шум); отношение высот пиков EtOH/i-PrOH: Y = 0.075; коэффициент регрессии и свободный член по калибровке: a = 0.694, B = -0.031. Вычислим концентрацию этанола по формуле: C = k(Y - В)/a = 0.95(0.075 – (-0.031))/0.694 ≈ 0.06 промилле. Если мы обратимся к шкале выраженности алкогольного опьянения Прозоровского и др., увидим, что «реципиент» был трезв как стеклышко. Большего от нас никто не требует. Если бы пик EtOH оказался вдвое выше, конечный результат (выраженность алкогольного опьянения) был бы точно таким же. Так в чем практический смысл количественной оценки сверхмалых концентраций, лежащих ниже границы заданной физиологической нормы? С уважением, ws_K. |
|
|
|
|
|
| Deminolog |
1.04.2013 - 21:07
Сообщение
#30 |
|
|
Здравствуйте!
Благодарю на добром слове, мне также приятно вести конструктивные беседы с Вами. Мои познания в статистическом анализе весьма ограничены, поскольку я предпочитаю сугубо практический подход к решению подавляющего большинства задач Начнем, пожалуй. Прежде всего внесу уточнение - я не эксперт и не токсиколог, химик - да О коэффициенте: я имел в виду коэффициент, позволяющий сделать поправку на матричные влияния. Матричное влияние - это не обязательно пик, который выходит рядом или вообще на том же времени, может проявляться следующая ситуация: фоновый сигнал при вколе становится другим, более интенсивным, нежели при вколе чистого соединения, например. И тут, чтобы не дать заниженный результат, вводится поправочный коэффициент, который будет иметь свое собственное значения для крови, другое значение для ткани, третье - для мочи... При этом он не имеет отношения к градуировке. Можно провести учет коэффициента другим образом: взять бланк (образец определенной биожидкости или ткани, в котором заведомо нет определяемого компонента, однако при этом сохраняется полностью матрица), добавить в него аналит и провести определение. Кстати, интересен также и такой момент: если раньше считалось, что введение внутреннего стандарта в биожидкость позволит учесть все влияния, то сейчас стараются предварительно выявлять влияние матрицы на каждый из определяемых компонентов, ибо он может разительно отличаться.Вот, собственно, о каком коэффициенте я и говорил. Думается мне, что мы говорим об одном и том же коэффициенте. По второму пункту: да, я имел в виду коэффициент b. Под значимым я понимал значение, отличное от нуля. Но даже когда b = 0 нельзя (никогда нельзя) говорить о том, что график начнется из нуля, поскольку всегда есть определенное значение шума, отличное от нуля. Таким образом, если концентрация аналита = 0, величина сигнала все равно нулю не равна, однако в то же время это не исключает того, что Ваш коэффициент b может быть равен нулю. Причиной тому, как раз, наш диапазон линейности, который является определенным интервалом. По отношению к приведенному примеру (5 и 6 промилле), разве кто-то запрещает добавить эти точки в градуировку? На мой взгляд, при необходимости точки можно добавить, либо почему бы не написать фразу "превышает 4 промилле"? Если этих точек нет, то это, пожалуй, самый честный ответ. То же касается точек, лежащих ниже. А что касается того, как бы поступил я... То тут, честно говоря, вижу единственный вариант - добавить точки, которые мне нужны, после чего оценю их вклад в градуировку. Тангенс угла наклона не изменился - великолепно, работаем. Изменился - выясню где, вероятно сделаю два диапазона линейности, если ошибка будет превышать 5% (мой личный критерий, кто-то может взять больше, кто-то меньше). Есть законы науки, которые для меня превыше методики, буду честен. Колонка, температурная программа, объем вкола, модель прибора и любые его надстройки - все это в публикациях достаточно давно называют условиями хроматографирования, а любая процедура, относящаяся к пробе - подготовка проб. Для меня эта терминология близка и привычна, поэтому я Вас не понял, простите. По поводу масс-спектрометрического и спектрофотометрического детекторов - эти детекторы используются в хроматографии. Второй используется только в ВЭЖХ. Мое высказывание по поводу градуировки по 2-м точкам было несколько эмоционально. Больше, чем следовало бы. Просто для меня неприемлема экстраполяция. Особенно в области низких концентраций, где любой вклад в ошибку становится существенным. Ну и касательно последнего примера: в Вашем случае да, определение следовых количеств не требуется. Однако существует множество задач, где это актуально и востребовано. Например, в допинг-контроле при определении соединений эндогенного происхождения. Это очень даже актуально, особенно, учитывая те пределы детектирования, которые ставит перед своими лабораториями ВАДА. Думаю, мне стоило уточнить раньше, что я говорю не только о задаче определения спирта, а точнее, не столько о нем, сколько о других задачах. Как-то так С уважением. |
|
|
|
|
|
|
| Сейчас: 12.07.2025 - 02:23 |
© 2002-2017 Форум судебных медиков России
Копирование материалов сайта возможно при условии размещения активной ссылки на источник.
Копирование материалов сайта возможно при условии размещения активной ссылки на источник.